Using alto on vaginal microbiome
data
microbiome-demo.Rmd
suppressPackageStartupMessages({
library(alto)
library(dplyr)
library(magrittr)
library(ggplot2)
library(tibble)
library(stringr)
library(tidyr)
})This document showcases the alto package functions
applied to vaginal microbiome data.
## Loading the data
We use the data published with the article “Replication and refinement of a vaginal microbial signature of preterm birth in two racially distinct cohorts of US women” by Calahan et al., 2017, PNAS.
A .zip file containing the related data can be downloaded from the Stanford data catalog.
For convenience, the data file (i.e. the tables contained in the
processed.rda file) has been attached to the
alto package as vm_data. For details, type
?vm_data.
names(vm_data)
#> [1] "sample_info" "counts" "taxonomy"First, we do some pre-processing of the data. Specifically, we change
the colnames to replace ASV DNA sequences by human-friendly
names built from the taxonomy table.
# we create the human-friendly ASV names
# by concatenating Genus and Species and an identifying number
# as a given species can be represented by several ASVs
vm_data$taxonomy <-
vm_data$taxonomy %>%
as.data.frame() %>%
group_by(Genus, Species) %>%
mutate(
ASV_name =
paste0(
ifelse(is.na(Genus),"[unknown Genus]", Genus), " (",
ifelse(is.na(Species),"?", Species), ") ",
row_number()
)
) %>%
ungroup()
# then, we replace the colnames of the count matrix by these new names.
# Note that the tax table rows are ordered as the count matrix columns,
# allowing us to assign without matching first.
colnames(vm_data$counts) <- vm_data$taxonomy$ASV_nameUsing alto on these data
Our first step is to run the lda models for varying number of topics,
i.e. from 1 to 18 topics. For this, we use the
run_lda_models function. This can take a while on a
personal computer. For example, it takes about 10 minutes on the
authors’ laptops.
lda_varying_params_lists <- list()
for (k in 1:18) {lda_varying_params_lists[[paste0("k",k)]] <- list(k = k)}
lda_models <-
run_lda_models(
data = vm_data$counts,
lda_varying_params_lists = lda_varying_params_lists,
lda_fixed_params_list = list(method = "VEM"),
dir = "microbiome_lda_models/",
reset = FALSE,
verbose = TRUE
)
#> fitting model k1
#> fitting model k2
#> fitting model k3
#> fitting model k4
#> fitting model k5
#> fitting model k6
#> fitting model k7
#> fitting model k8
#> fitting model k9
#> fitting model k10
#> fitting model k11
#> fitting model k12
#> fitting model k13
#> fitting model k14
#> fitting model k15
#> fitting model k16
#> fitting model k17
#> fitting model k18We can now align the topics from each consecutive models:
aligned_topics_product <-
align_topics(
models = lda_models,
method = "product")
aligned_topics_transport <-
align_topics(
models = lda_models,
method = "transport") Alignement visualization
The alignments can be visualized with the plot
function.
plot(aligned_topics_product)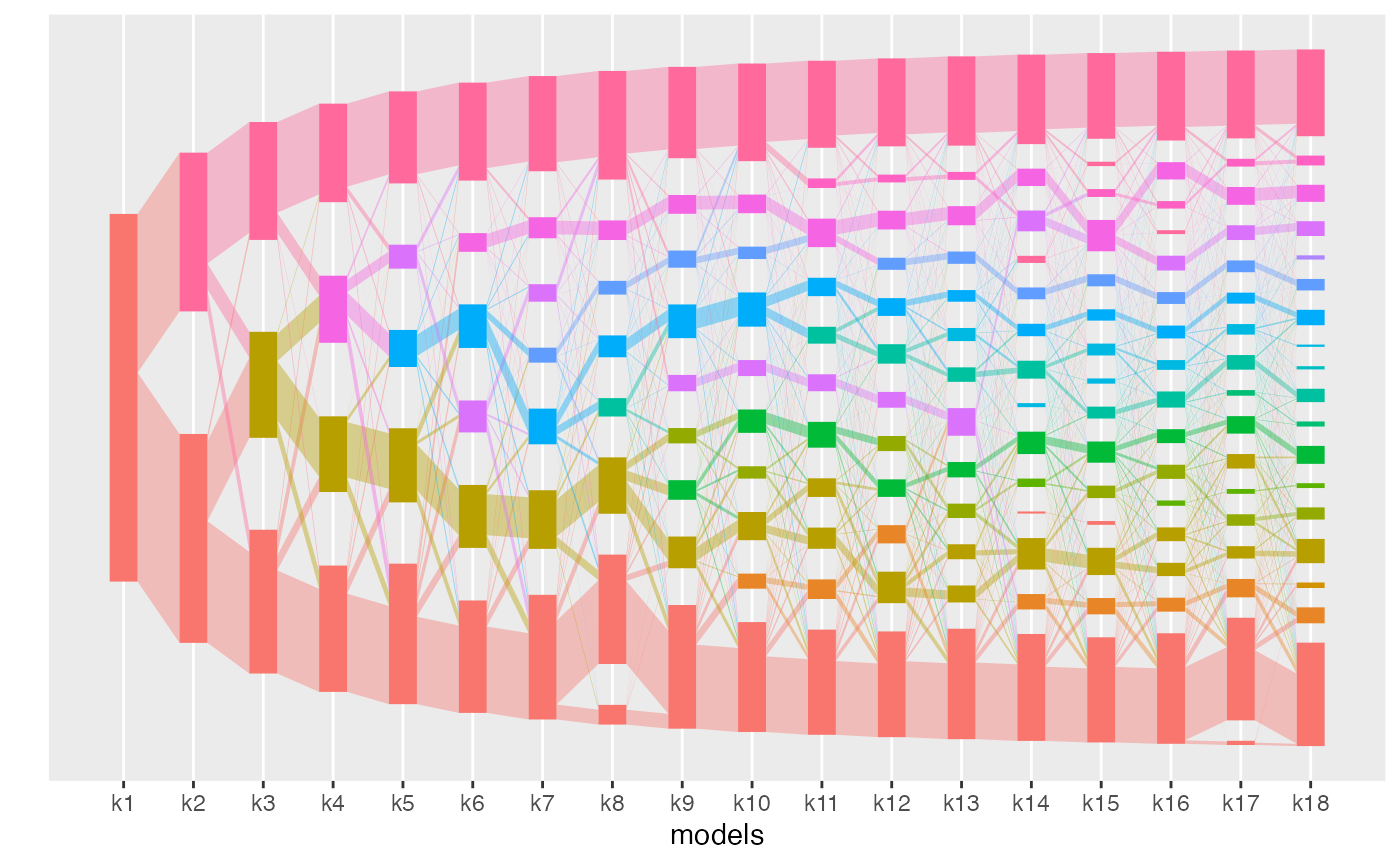
plot(aligned_topics_transport)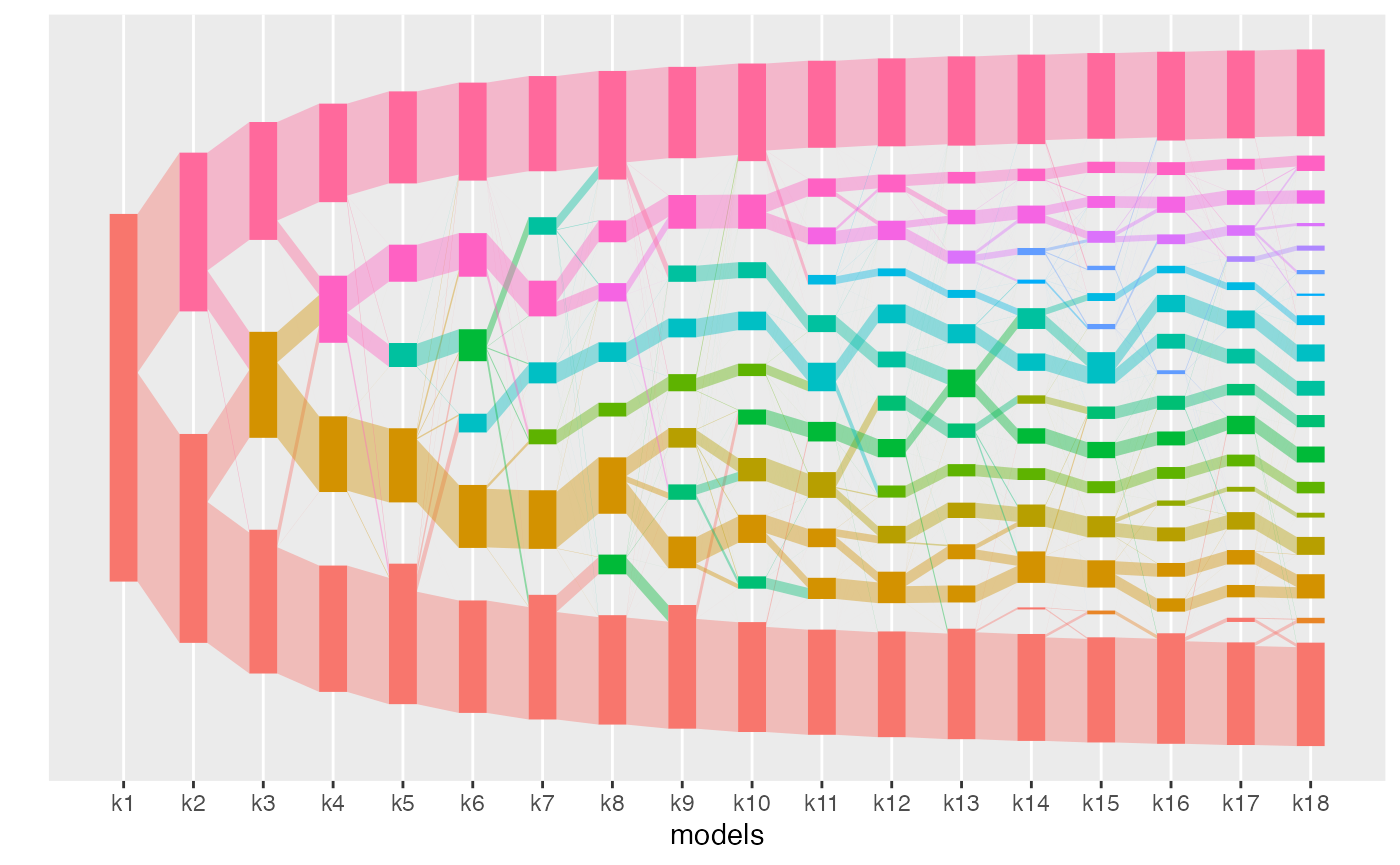
Key number of topics
The number of key topics is computed with the function
compute_number_of_key_topics. The function
plot_number_of_key_topics can then be used to visualize
that number across models.
compute_number_of_paths(aligned_topics_product) %>%
plot_number_of_paths() +
ggtitle("Method: product")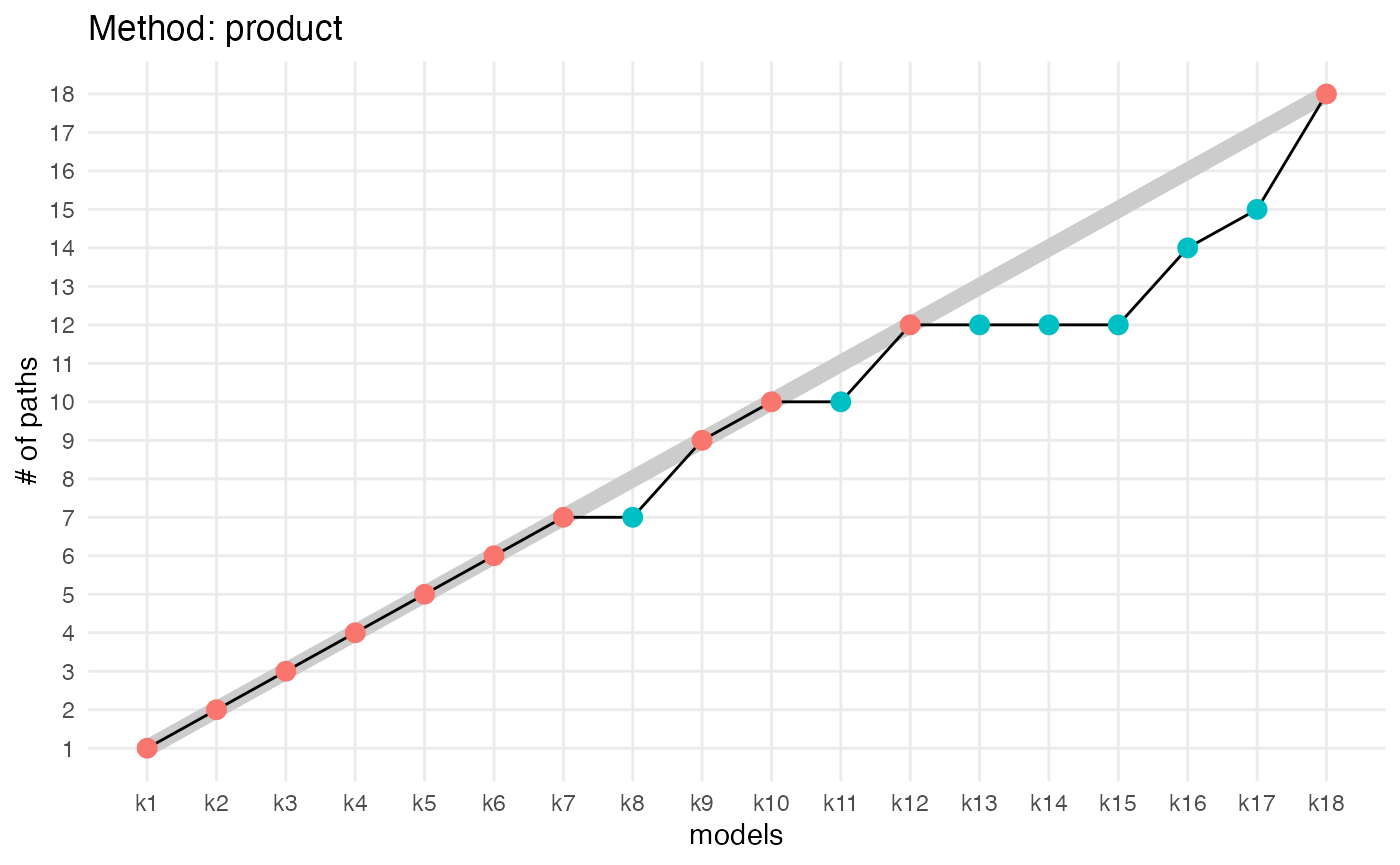
compute_number_of_paths(aligned_topics_transport) %>%
plot_number_of_paths() +
ggtitle("Method: transport")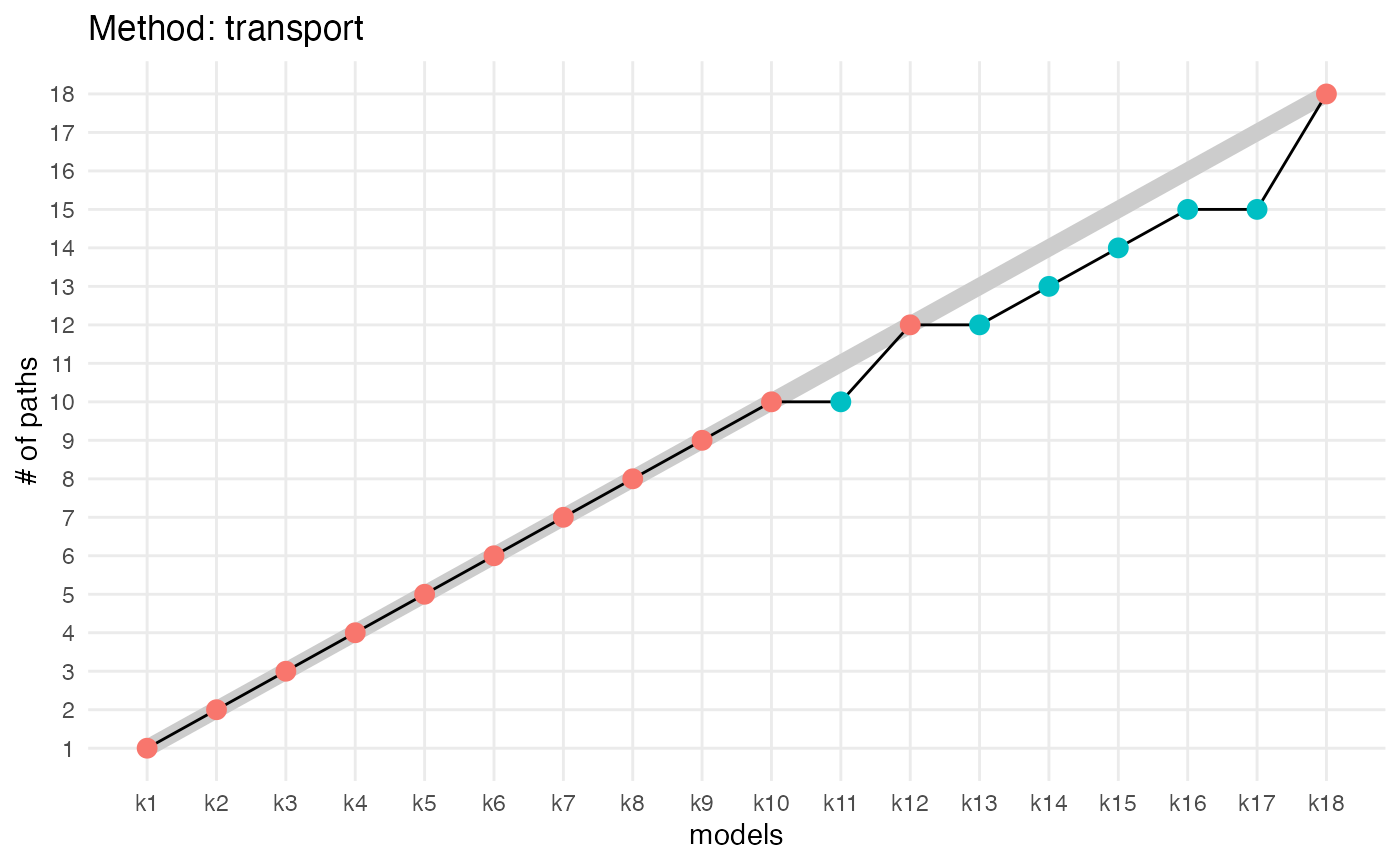
The number of key topics shows a small plateau around K = 12 with both methods (product and transport). As in the simulations, the number of key topics are lower and the plateau is stronger when key topics are identified by the product than by the transport alignment. The fact that the plateau is small is likely indicative that the data generation process does not strictly follow the LDA model assumption.
Note: for convenience, the plot function includes
options for labeling the topics with their path ID, and with their
composition. Type ?plot_alignment for details about these
options.
aligned_topics_transport_12 <- align_topics(lda_models[1:12], method = "transport")
plot(aligned_topics_transport_12, add_leaves = TRUE, label_topics = TRUE)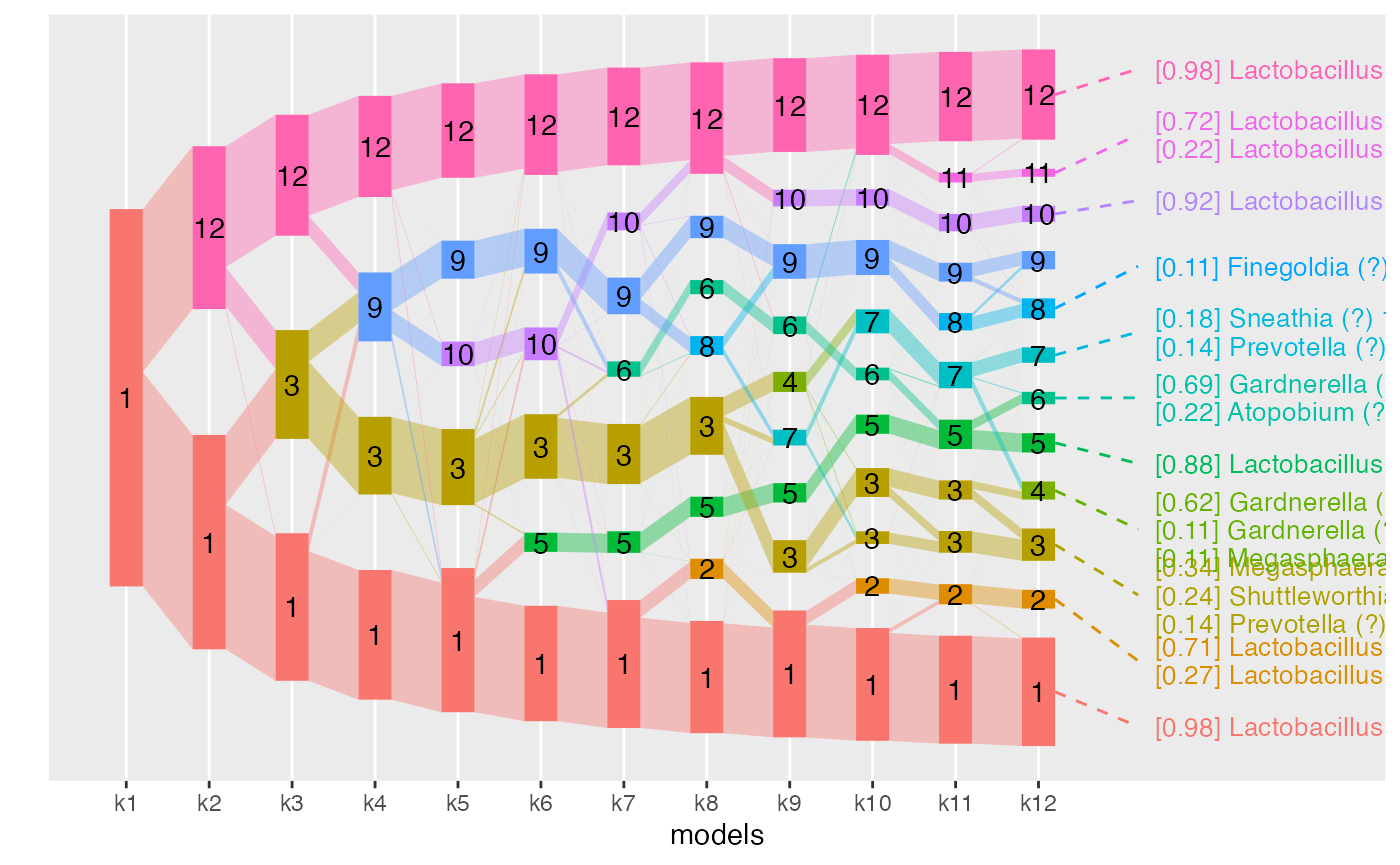
We can also evaluate the coherence and ancestry scores of these
topics with the plot function and the color_by
option.
Alignment with diagnostics scores
plot(aligned_topics_product, color_by = "coherence")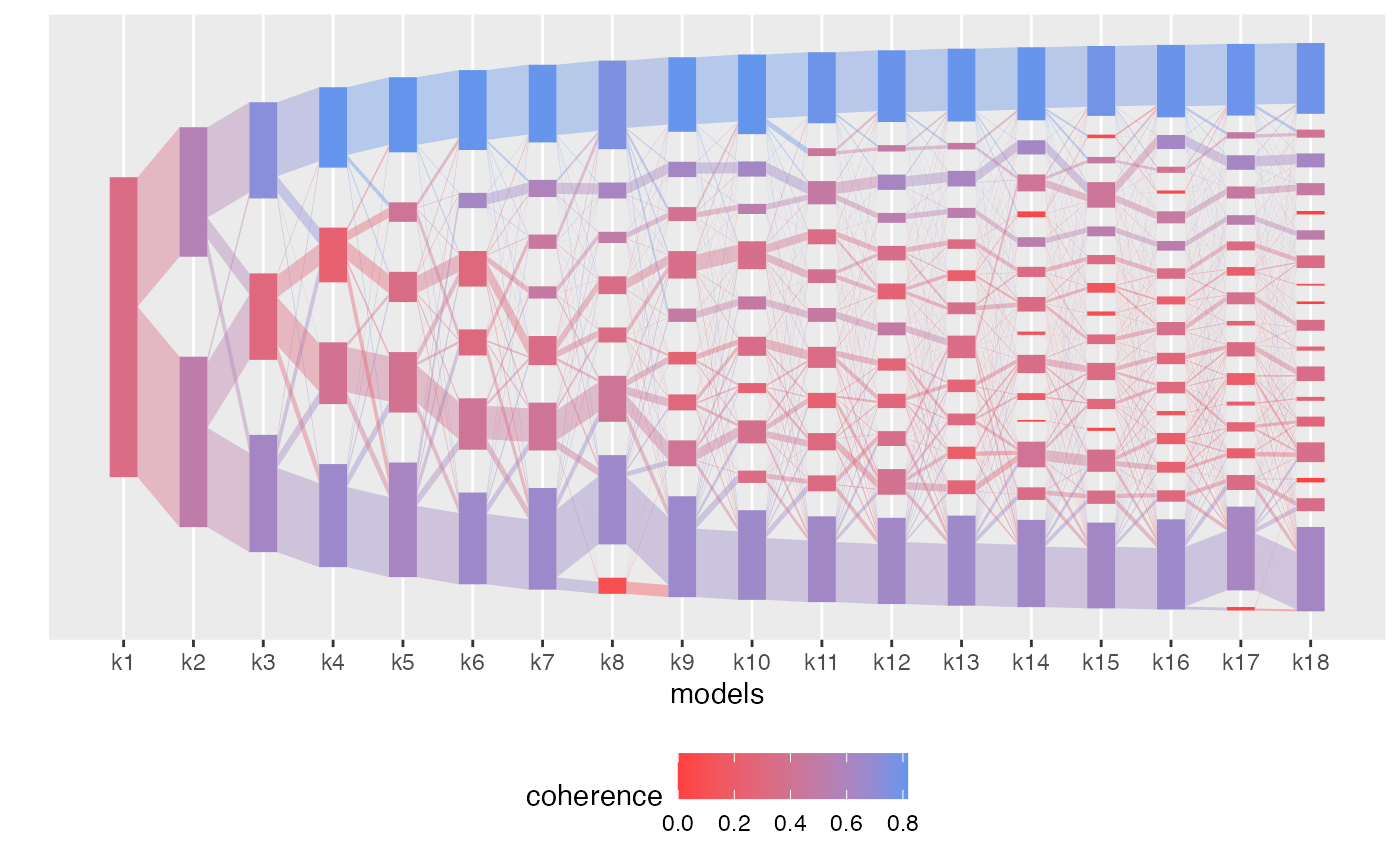
plot(aligned_topics_transport, color_by = "coherence")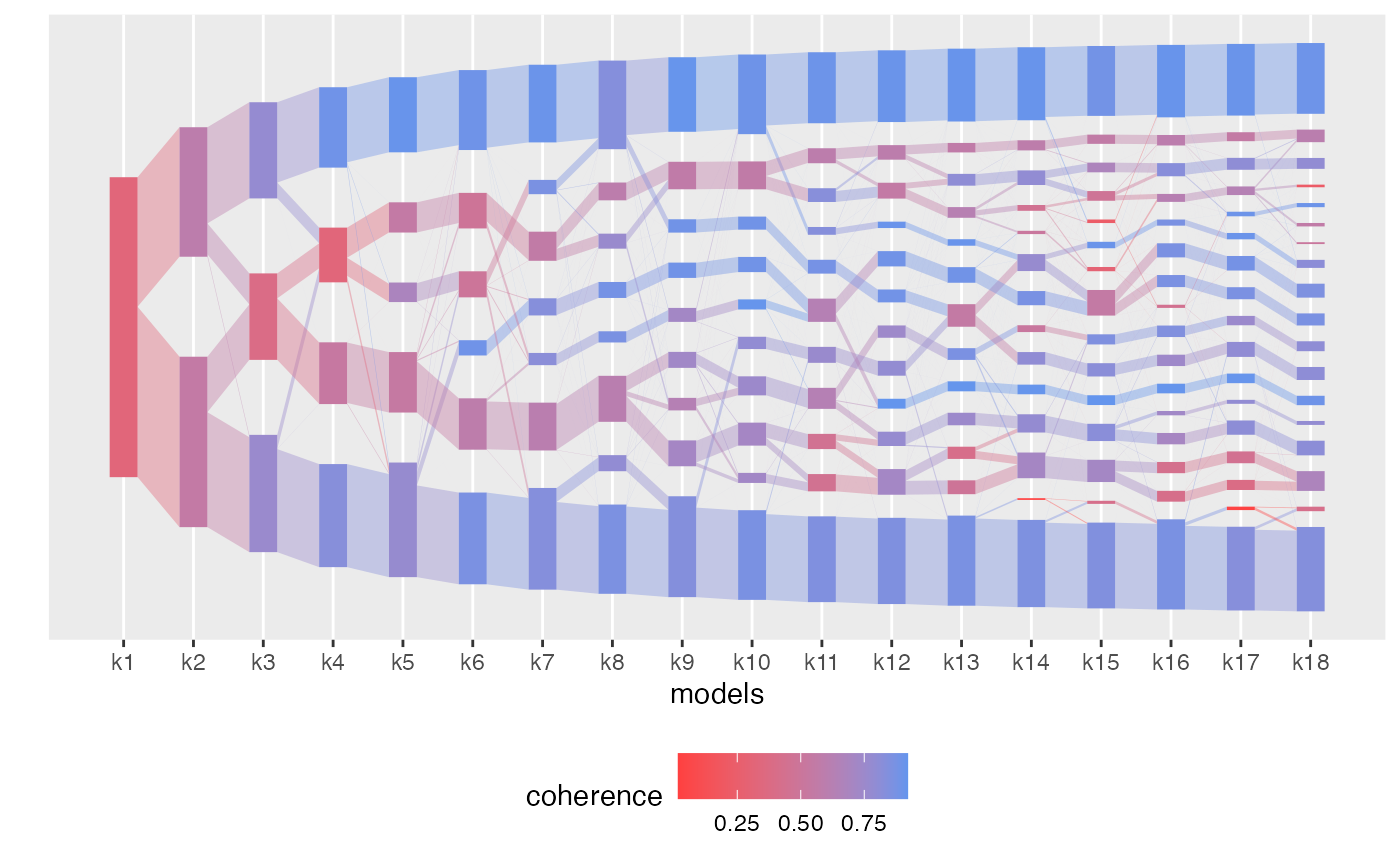
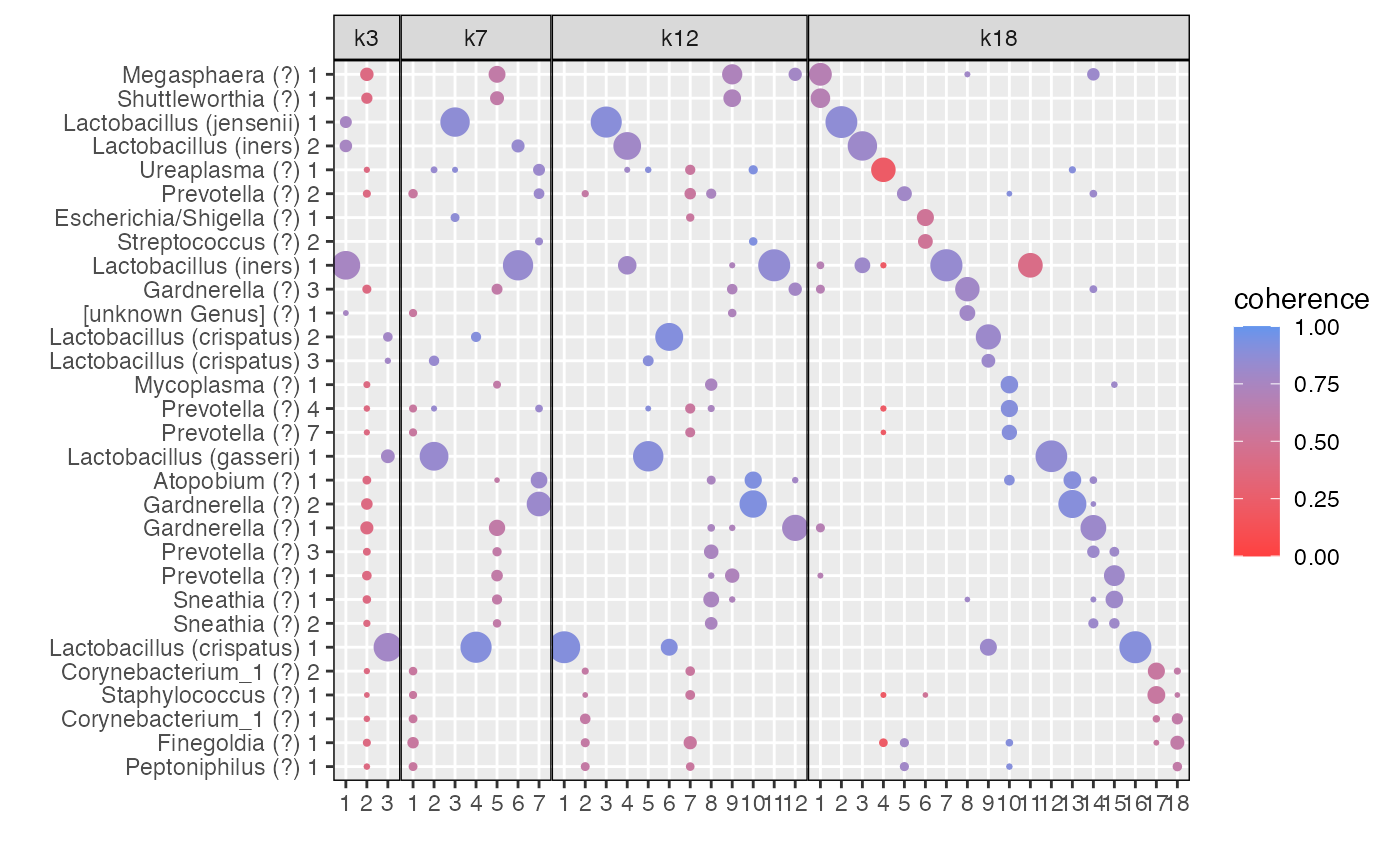
Most of the identified topics around \(K=12\) are coherent across \(K\).
plot(aligned_topics_product, color_by = "refinement")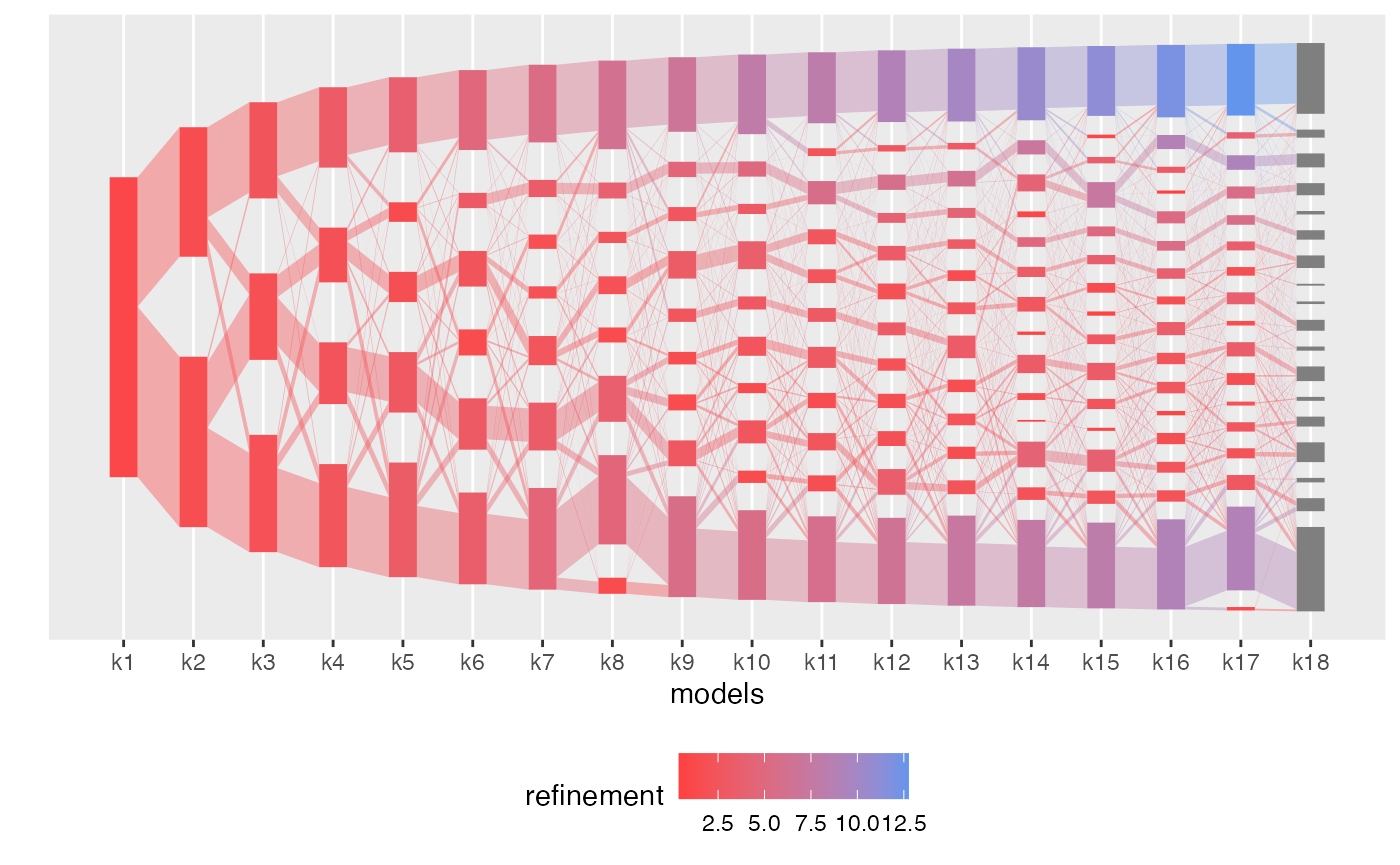
plot(aligned_topics_transport, color_by = "refinement")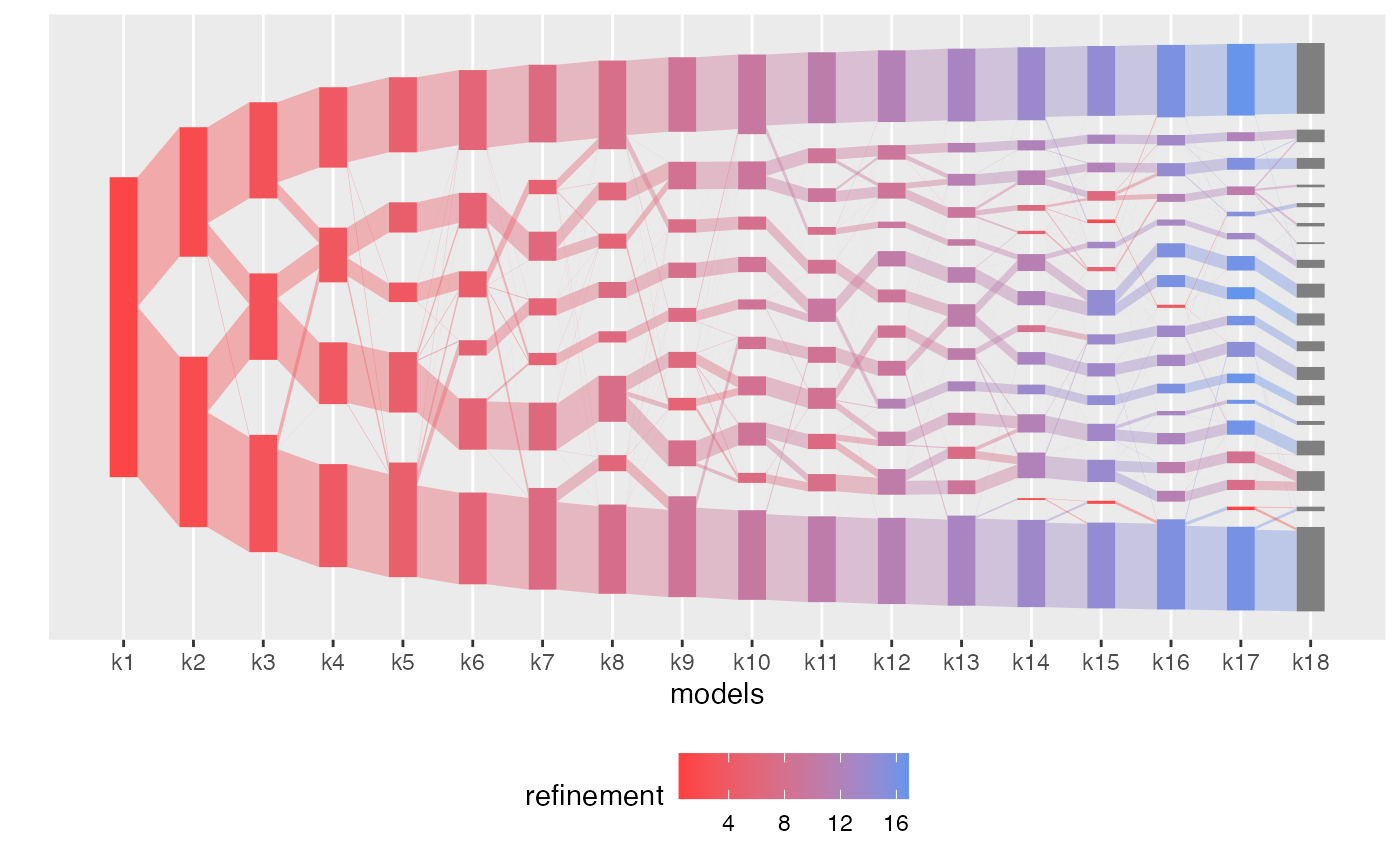
Interestingly, by \(K=7\), the identified topics correspond to the four Lactobacillus community state types (CST) and to three non-Lactobacillus topics. Among these, one topic, dominated by a specific strain of Gardnerella and Atopobium, remains coherent across models. The two remaining topics at \(K=7\) have little overlap and high refinement scores, indicative that these topics successfully identify two distinct groups of communities, which are revealed as robust topics as \(K\) is increased.
The function plot_beta allows to visualize the betas for
a selection (or all) models.
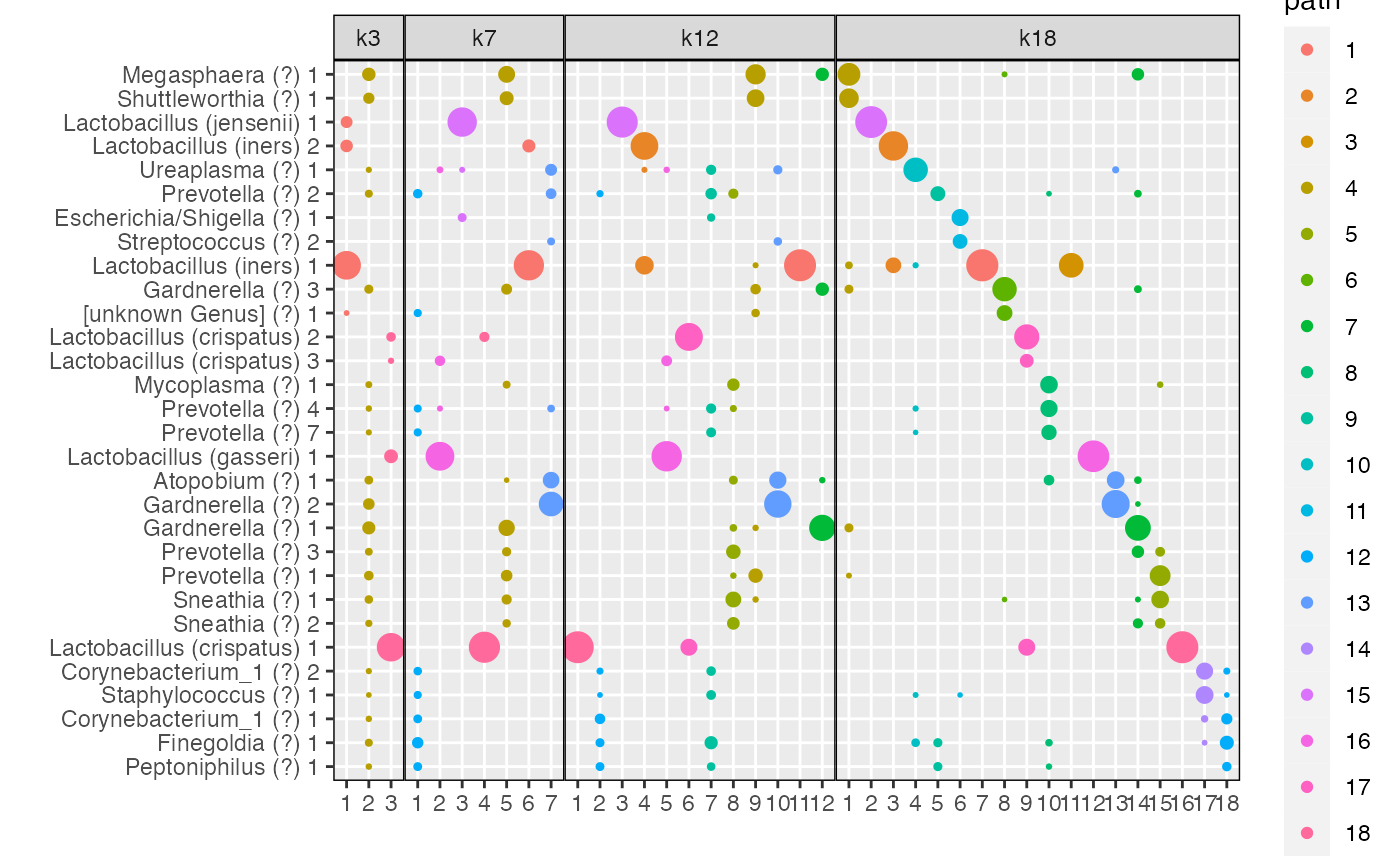
By \(K=18\) topics are sparse and share little overlap between them (Fig \(\ref{fig:microbiome_figure}\)d), which may reflect over-fitting.
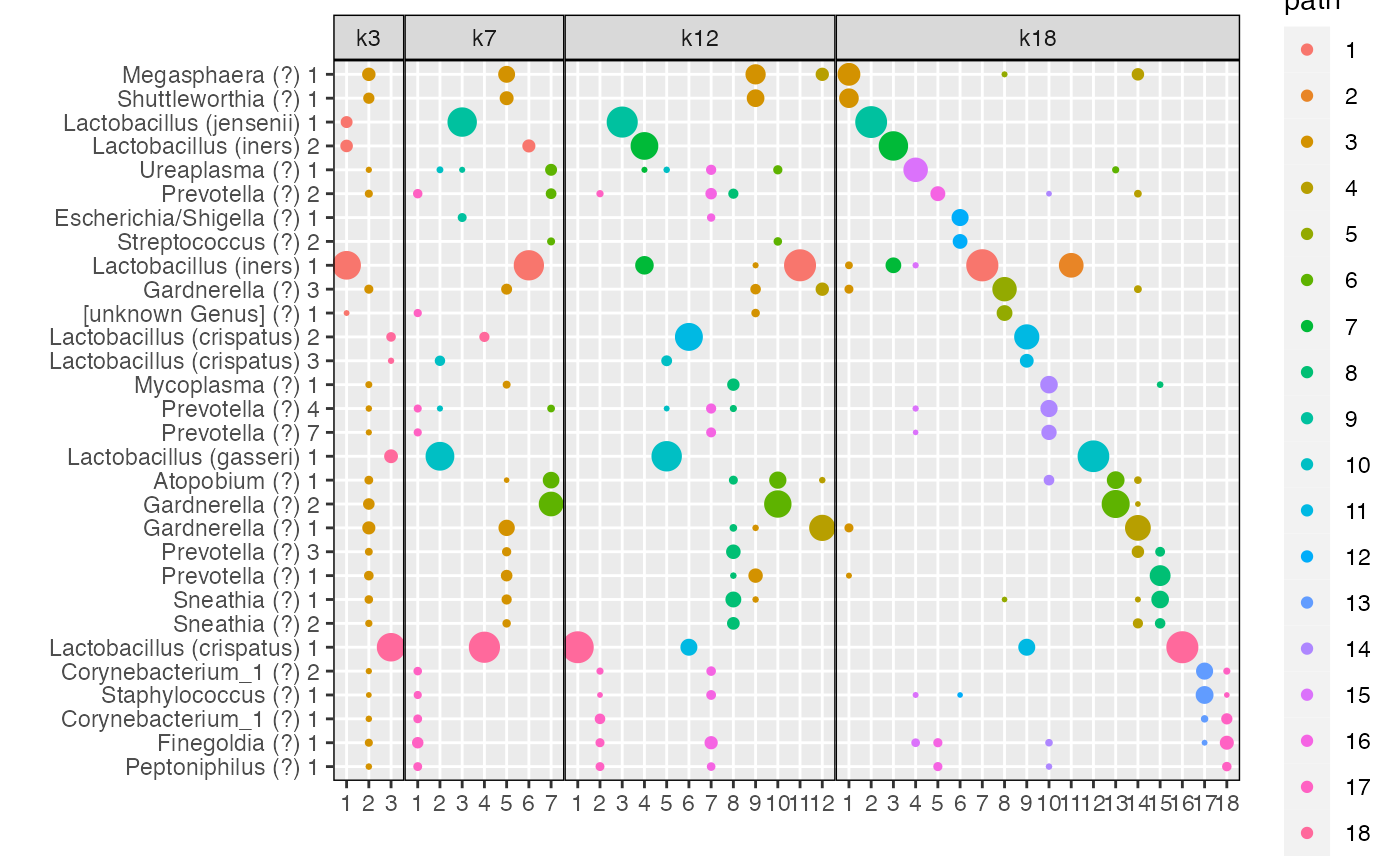
The plot_beta function also has a color_by
option, which allows to visualize the coherence of topics.
plot_beta(aligned_topics_transport,
models = c("k3", "k7","k12","k18"),
threshold = 0.005,
color_by = "coherence")